Marcações
Pedir Consulta/Pedir Exame – Após preencher os respetivos formulários, estes pedidos serão tratados pelos nossos serviços e receberá a confirmação por e-mail. As indicações sobre os Acordos (Seguro/Subsistema) são meramente informativas, nesse sentido peça confirmação no campo de “Observações”.
Marcar Consulta/Exame myHPA – Ao aceder à myHPA Saúde pode efetuar a marcação em tempo real, diretamente na agenda do Médico mediante a disponibilidade que o mesmo apresenta.
Pode ainda contactar-nos através do 282 420 400 Algarve | 269 630 370 Alentejo | 291 003 300 Madeira
Horário serviço de marcação/atendimento por telefone:
Segunda a Sexta: 08h às 19h, Sábado: 08h às 17h, Domingo e Feriados: Encerrado
(Chamada para a rede fixa nacional)
O seu pedido foi recebido com sucesso. Iremos confirmar a sua marcação em breve.
Descarregue a myHPA Saúde:


NEWSLETTER
Subscreva e fique a saber em primeira mão todas as novidades, campanhas e rastreios gratuitos.
Dr. João Costa
Anatomopatologista

O papel da Anatomia Patológica na deteção da Síndrome de Lynch
HPA Magazine 24 // 2025
A síndrome de Lynch é uma síndrome de predisposição hereditária ao cancro, associada a alterações germinativas envolvendo os genes MLH1, PMS2, MSH2, MSH6 e EPCAM (entre outros), e está associada a um elevado risco de diversas neoplasias, principalmente carcinoma colorretal e do endométrio. Está também associada a um risco aumentado de carcinomas do ovário, urotelial, estômago, intestino delgado, pâncreas, trato hepatobiliopancreático, neoplasias do sistema nervoso central e pele, entre outras. É estimado que cerca de 1 em cada 450 pessoas têm síndrome de Lynch e que apenas cerca de 5% dessas pessoas são diagnosticadas. O risco cumulativo ao longo da vida de carcinoma colorretal e de carcinoma endometrial em doentes com esta síndrome é cerca de 10-80% e 21-71%, respetivamente (dependendo do gene alterado), e estima-se que 3-5% dos carcinomas colorretais e 2-6% dos carcinomas endometriais estão associados a esta síndrome. A deteção da síndrome de Lynch tem extrema importância, não só porque os doentes beneficiam de protocolos de vigilância e terapias preventivas adaptadas, mas também para detetar outros portadores assintomáticos nas famílias desses doentes.
Os genes MLH1, PMS2, MSH2, MSH6 estão envolvidos no mecanismo de mismatch repair. Este mecanismo é responsável pela reparação de um tipo específico de erros na replicação do ADN. Defeitos neste mecanismo levam à acumulação destes erros, especialmente em microssatélites (pequenas sequências repetitivas no ADN), levando à instabilidade de microssatélites.
Esta síndrome tem um padrão de hereditariedade autossómica dominante. O doente herda um alelo com um gene afetado de um dos progenitores, sendo, no entanto, necessária a inativação do segundo alelo para ocorrer carcinogénese. A inativação do segundo alelo pode ocorrer por vários mecanismos, sendo o mais frequente, a hipermetilação do promotor do MLH1.
Os critérios clínicos para identificar doentes com alta probabilidade de síndrome de Lynch (critérios de Amsterdão, Bethesda e Bethesda modificados, entre outros) falham na identificação de um número significativo de doentes, pelo que novas estratégias de rastreio são necessárias. Atualmente, a National Comprehensive Cancer Network, a European Society For Medical Oncology e outras sociedades internacionais recomendam o rastreio universal de todos os casos de carcinomas colorretais e endometriais. A National Comprehensive Cancer Network recomenda também considerar o rastreio de todas as neoplasias sebáceas, bem como todos os carcinomas do intestino delgado, gástricos, ovário, uroteliais e adrenocorticais, e neoplasias do sistema nervoso central, independentemente da idade. Este rastreio é feito pelos anatomopatologistas com recurso a dois métodos: estudo imuno-histoquímico para detetar as proteínas envolvidas no mecanismo de mismatch repair, ou pesquisa de instabilidade de microssatélites, ambas no tecido tumoral. Para além de detetar doentes em risco de serem portadores da síndrome de Lynch, este rastreio também dá informações adicionais, de carácter diagnóstico (estas técnicas são utilizadas pelos anatomopatologistas na avaliação de neoplasias, para fins de diagnóstico diferencial, e, nos casos dos carcinomas do endométrio e alguns tipos de carcinomas do ovário, para proceder à classificação molecular utilizando o algoritmo baseado no The Cancer Genome Atlas), prognóstico e teragonóstico (resposta à terapia com inibidores dos checkpoints imunes).
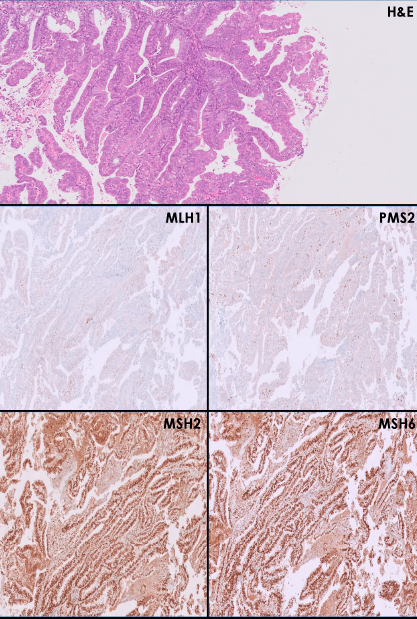
O estudo imuno-histoquímico deteta a expressão das proteínas codificadas pelos genes MLH1, PMS2, MSH2 e MSH6 no tecido tumoral. Num teste sem alterações é detetada a expressão normal de todas as 4 proteínas, sendo muito pouco provável que exista uma alteração patogénica num dos genes envolvidos. Já num teste com alterações, uma ou mais das proteínas não foi/foram detetada(s) e uma alteração patogénica do(s) gene(s) envolvido(s) pode estar presente, sendo recomendado fazer testes subsequentes para determinar se o doente é portador da síndrome de Lynch. O estudo imuno-histoquímico é mais barato, exige menos recursos, tem esquemas de controlo de qualidade externos melhor desenvolvidos, permite a correlação com os aspetos morfológicos do tumor e os resultados permitem guiar, de uma forma mais dirigida, os testes subsequentes necessários para despiste da síndrome de Lynch. Caso seja detetada a perda conjunta de MSH2 e MSH6, ou perdas isoladas de MSH6 e PMS2, o doente deve ser avaliado por Genética Médica para despiste da síndrome. Contudo, se ocorrer perda conjunta das proteínas MLH1 e PMS2, deve proceder-se, primeiro, nos casos de carcinoma colorretal, à pesquisa de hipermetilação do promotor do gene MLH1 e da mutação BRAF V600E, e, nos de carcinoma endometrial, à pesquisa isolada de hipermetilação do promotor do gene MLH1. Se estes testes forem negativos, o doente deve ser avaliado por Genética Médica para despiste da síndrome de Lynch (a presença de hipermetilação do promotor do gene MLH1 e da mutação BRAF V600E está associada a uma probabilidade muito baixa da síndrome referida).
A pesquisa de instabilidade de microssatélites é realizada através da avaliação de diferenças no comprimento de um conjunto de microssatélites, seja por polymerize chain reaction, seja por massively parallel sequencing. Este teste é mais caro, exige mais recursos, não permite a correlação com a morfologia e a sensibilidade dos diferentes testes disponíveis é variável, dependendo do tipo de cancro. Assim, a pesquisa de instabilidade de microssatélites tende a ser empregue em contextos mais específicos, nomeadamente, em situações em que o estudo imuno-histoquímico apresenta resultados de difícil interpretação, em casos sem perda de expressão das proteínas envolvidas no mecanismo de mismatch repair de doentes com alta probabilidade de serem portadores da síndrome de Lynch, e em situações de resultados contraditórios entre o estudo imuno-histoquímico e o estudo melecular. Em casos de tumores classificados como tendo instabilidade microssatélites alta (MSI high) ou resultados indeterminados, o doente deve ser avaliado por Genética Médica para despiste da síndrome de Lynch. Mais de 90% dos tumores em doentes com síndrome de Lynch apresentam instabilidade de microssatélites /ausência de imunoexpressão de uma ou mais proteínas – caso a suspeita clínica para esta síndrome seja elevada, deve-se proceder à pesquisa de mutações germinativas, mesmo que o tumor seja negativo. A taxa de concordância entre o estudo imuno-histoquímico e pesquisa de instabilidade de microssatélites é de 95%, podendo optar-se por uma ou a outra modalidade, dependendo dos recursos locais. Existe uma taxa de falsos negativos de 5-10% com estudo imuno-histoquímico e de 5-15% com pesquisa de instabilidade de microssatélites.
Figura 1:
Um carcinoma endometrióide do endométrio, de baixo grau (bem diferenciado – G1), com perda de expressão das proteínas MLH1 e PMS2 e expressão mantida das proteínas MSH2 e MSH6. Este resultado é compatível com deficiência nos mecanismos de mismatch repair. Nestes casos, deve ser efetuada a pesquisa de hipermetilação do promotor do gene MLH1.